| Team Members: | Trevor V. Adriaanse1, Meshach Hopkins2, Rebecca Rachan3, and Subodh R. Selukar4 |
| Graduate Assistant: | Elias Al-Najjar5 |
| Faculty Mentor: | Kofi P. Adragni5 |
| Client: | Nusrat Jahan6 |
1Department of Mathematics, Bucknell University,
2Department of Computer Science and Electrical Engineering, University of Maryland, Baltimore County,
3Department of Mathematics, North Central College,
4Department of Biostatics and Biology, University of North Carolina, Chapel Hill,
5Department of Mathematics and Statistics, University of Maryland, Baltimore County,
6James Madison University

About the Team
Trevor V. Adriaanse is a rising junior at Bucknell University in Lewisburg, PA majoring in mathematics and minoring in Linguistics. Meshach Hopkins is a rising sophomore at UMBC majoring in Computer Engineering. Rebecca Rachan is a rising senior at North Central College in Naperville, IL majoring in mathematics. Subodh R. Selukar is a rising senior at UNC-CH double majoring in Quantitative Biology and Biostatistics. Team 3 worked with Graduate Assistant Elias Al-Najjar and Faculty Mentor Kofi Adragni. Our client, Nusrat Jahan, is a Statistics Professor at James Madison University. In our project, we aimed to find a set of genes to best predict Alzheimer’s Disease.
Predicting Alzheimer’s Disease
Alzheimer’s Disease (AD) is a progressive, degenerative neurological disorder chiefly present in the elderly. The disease begins in the hippocampus, where irregular protein fragments called plaques and tangles slowly destroy brain cells. We were provided microarray data from the Hisayama study that includes a listing of approximately 32,000 gene expressions from 79 postmortem patients, 32 of which had AD. Our primary goal is to find a relationship between gene expression and presence of AD. We implement a three step statistical proceedure: screening, sparse sufficient dimension reduction (SDR), and hierarchical clustering. Further, we parallelize the existing R code to enhance execution speed and conduct a performace analysis.
Methodology
We begin with an array X of gene expressions and Y the response variable, where Y=0 if AD is not present and Y=1 if AD is present. We use an inverse regression approach because of the data sampling scheme. The high number of gene expressions and small sample size motivates a SDR. A reduction is sufficient if we can replace X with a lower dimensional function, still containing all relevant information about the response. To implement the dimenstion reduction we use an eigenvalue method called Principal Fitted Components (PFC) Model. This yields parameters that give the relationship between AD and the genes, as well as the interdependence of genes.
Our method is as follows:
- Screen data with t-tests.
- Perform sparse estimations and cross-validation
- Cluster genes exhibiting mutual dependence
Implementation
We adapted the previous works of Dr. Adragni to implement our methodology in R. We parallelized our main function on the maya 2013 cluster using the snow package in R. We ran simulations as a performance analysis. Finally, we applied our code to the AD data set.
Results
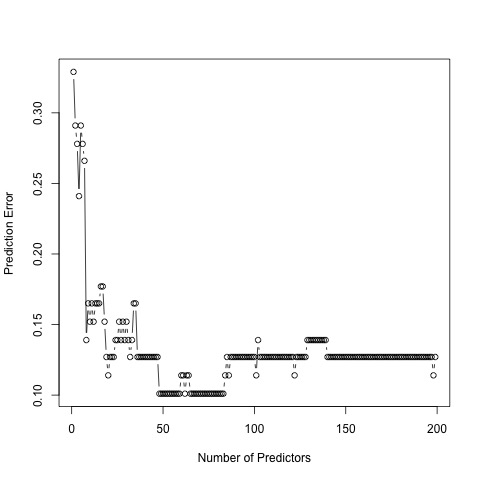
Prediction error for the 200 most significant genes.
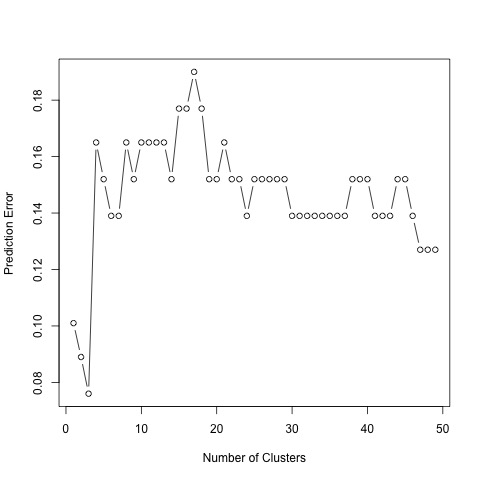
Prediction error using the 49 genes that best predict AD.
To compute the prediction error in both the sparse estimation and clustering steps, we used a cross-validation method. We want to have a low prediction error in our results. As seen, the prediction error is minimized for 49 predictors. Hence, our findings indicate 49 significant genes to best predict AD. In the second figure, prediction error is minimized for 3 clusters. Therefore, we group our 49 genes into 3 groups, specified by our clustering structure.
Parallelization
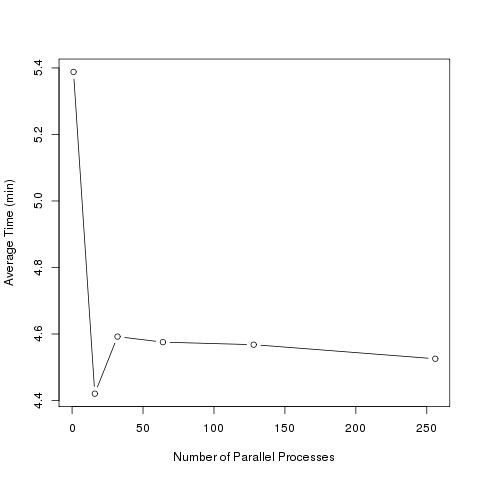
Performance study of our main R function
We experimented with parallelizing various subfunctions. The above graph illustrates the performance study on the most important of these functions. Our results show parallelization improved performance speed most substantially using one node with sixteen processes per node.
Conclusions
We found 49 genes grouped into 3 clusters to predict Alzheimer’s Disease. We aim to extend our research by comparing our model with a logistic regression model to determine the efficacy of our methodology. Further, we will examine the biological relevance of our findings by identifying the genes, their biological functions, and their relationship to AD. We hope that our findings will result inclinical significance by providing better prediction of Alzheimer’s.
Links
Trevor V. Adriaanse, Meshach Hopkins, Rebecca Rachan, Subodh R. Selukar, Elias Al-Najjar, Kofi P. Adragni, and Nusrat Jahan. Statistical analysis of a case-control Alzheimer’s disease: a retrospective approach with sufficient dimension reduction. Technical Report HPCF-2015-23, UMBC High Performance Computing Facility, University of Maryland, Baltimore County, 2015. (HPCF machines used: maya.). Reprint in HPCF publications list
Poster presented at the Summer Undergraduate Research Fest (SURF)
Click here to view Team 1’s project
Click here to view Team 2’s project
Click here to view Team 4’s project
Click here to view Team 5’s project
Click here to view Team 6’s project
Click here to view Team 7’s project
Click here to view Team 8’s project